بیماری گیرن باره چیست؟
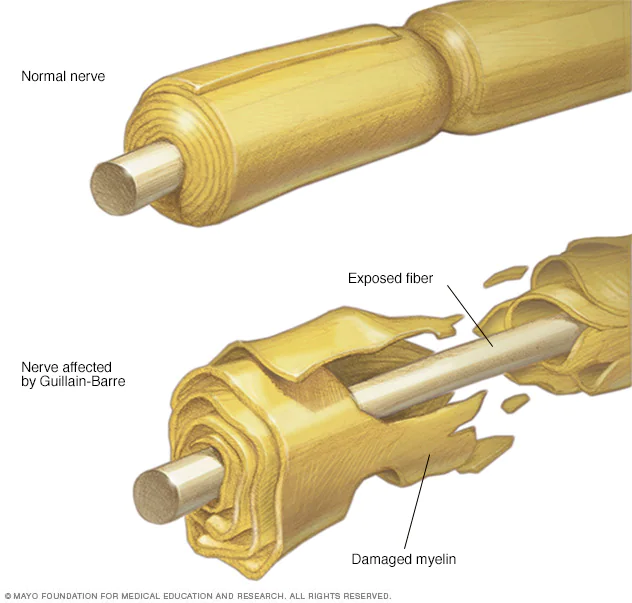
خلاصه مقاله
گیلن باره یک بیماری خطرناک پیشرنده است که
#با_مقاله_درس_بخوانیم
#Guillain_Barre_Syndrome
واکسن ها و سندروم گیلن-باره
افزایش نظارت بر سندرم گیلن باره مرتبط با تجویز واکسن به دلیل گزارشهایی مبنی بر افزایش خطر ابتلا به سندرم گیلن باره (تقریباً یک مورد از هر 100000 واکسن) در افرادی که واکسن آنفولانزای H1N1 در سال 1976 دریافت کرده بودند، انجام شد.
البته واکسیناسیون خطر ابتلا به عود را در افرادی که قبلاً به سندرم گیلن باره مبتلا شده بودند افزایش نداد.
Immune checkpoint inhibitors و سندروم گیلن-باره
با معرفی مهارکننده های ایمنی به عنوان دارو در درمان سرطان، سرطان هایی که قبلا غیر قابل درمان بودند، اکنون بهبود یافته اند. عوارض جانبی عصبی، هرچند نادر، گزارش شده است، از جمله وضعیتی شبیه به سندرم گیلن باره.
میانگین زمان شروع در بیماران مبتلا به سندرم گیلن باره معمولاً پس از سه دوره درمان با مهارکننده های ایمونولوژیک بود و پیشرفت بیماری سریع بود.
توصیه درمانی کنونی برای عوارض عصبی ناشی از درمان با مهارکننده های ایمونوتیک، توقف داروی مسبب و شروع مصرف استروئیدها است.
با این حال، از آنجایی که به نظر می رسد سیر بالینی سندرم گیلن باره ناشی از مهارکننده های ایمنی شبیه به سندرم کلاسیک گیلن باره است، ایمونوگلوبولین وریدی یا تبادل پلاسما باید در نظر گرفته شود.
نقش آنتیبادیها
گانگلیوزیدها گلیکولیپیدهای حاوی اسید سیالیک هستند که در سیستم عصبی پستانداران، به ویژه در گره های Ranvier و پایانه های عصبی حرکتی غنی شده اند. اهمیت آنها به عنوان هدف در موشهای تراریخته نشان داده شده است که گانگلیوزیدهای پیچیده را منحصراً در نورونها بیان میکنند. مکانیسم اساسی برای نوروپاتی با واسطه آنتیبادی شامل تعدیل عملکرد کانال یونی در گرههای Ranvier، سمیت سلولی وابسته به مکمل در گرهها و پایانههای عصبی حرکتی، و تداخل با بازسازی عصبی.
زیرگروه های سندرم گیلن باره اغلب با آنتی بادی های آنتی گانگلیوزید خاص همراه هستند که نشان دهنده غنی سازی نامتناسب گلیکولیپیدهای هدف در اعصاب مختلف است.
معیارهای تشخیصی
معیارهای متعددی برای کمک به پزشکان در تشخیص سندرم گیلن باره ایجاد شده است.
حداقل، تشخیص سندرم گیلن باره مستلزم وجود ضعف متقارن شل و کاهش رفلکس در غیاب علل جایگزین است.
معیارهای برایتون همچنین تعریف مورد جداگانه ای را برای سندرم میلر فیشر در نظر می گیرد، که نیاز به حضور سه گانه بالینی افتالمپلژی دو طرفه، کاهش رفلکس ها و آتاکسی، همراه با فقدان ضعف اندام و درگیری CNS برای تحقق یک قطعیت تشخیصی سطح 3 دارد.
دستیابی به قطعیت های تشخیصی بالاتر در هر دو سندرم گیلن باره و سندرم میلر فیشر مستلزم وجود یک بیماری تک فازی است که در عرض 28 روز به حد نادر می رسد، تجزیه آلبومین سیتولوژیکی مایع مغزی نخاعی و شواهد الکترودیاگنوستیک نوروپاتی.
در عمل، ویژگی های بالینی سندرم گیلن باره متغیر است. اگرچه در هیچ یک از معیارهای تشخیصی گنجانده نشده است، یک بیماری پیشین در 4 هفته قبل در 76٪ از بیماران وجود دارد.
الگوی ضعف در سندرم گیلن باره به اندام ها نیز محدود نمی شود و می تواند شامل عضلات عصب دهی شده جمجمه، عضلات تنفسی و درگیری اتونوم شود. به ندرت، این الگوهای غیر معمول می تواند اولین تظاهر سندرم گیلن باره باشد.
امروزه هم از معیارهای تشخیصی برایتون و هم معیارهای موسسه ملی اختلالات عصبی و سکته مغزی ایالات متحده (NINDS) استفاده میشود.
گیلن باره در کودکان
تشخیص سندرم گیلن باره در کودکان می تواند چالش برانگیز باشد. با این حال، درد قابل توجهی در ارتباط با سندرم گیلن باره کودکان وجود دارد که می تواند ضعف اندام را بپوشاند و باعث تاخیر در تشخیص شود. هنگامی که مطالعات هدایت عصبی در کودکان قابل تحمل نباشد، تصویربرداری عصبی با MRI یا اولتراسوند می تواند تشخیص را تسهیل کند. کودکان مبتلا به سندرم گیلن باره پیش آگهی خوبی دارند، اما از آنجایی که گزارش هایی از مرگ و میر ناشی از اختلال عملکرد اتونوم وجود دارد، استراتژی های درمانی، همانطور که در سندرم گیلن باره بزرگسالان توصیه می شود، توصیه می شود.
گردآورنده: ساغر مومنی
ادیت:دکتر فرزان فهیم
🆔 @Neurosurgery_association
مقالات مرتبط
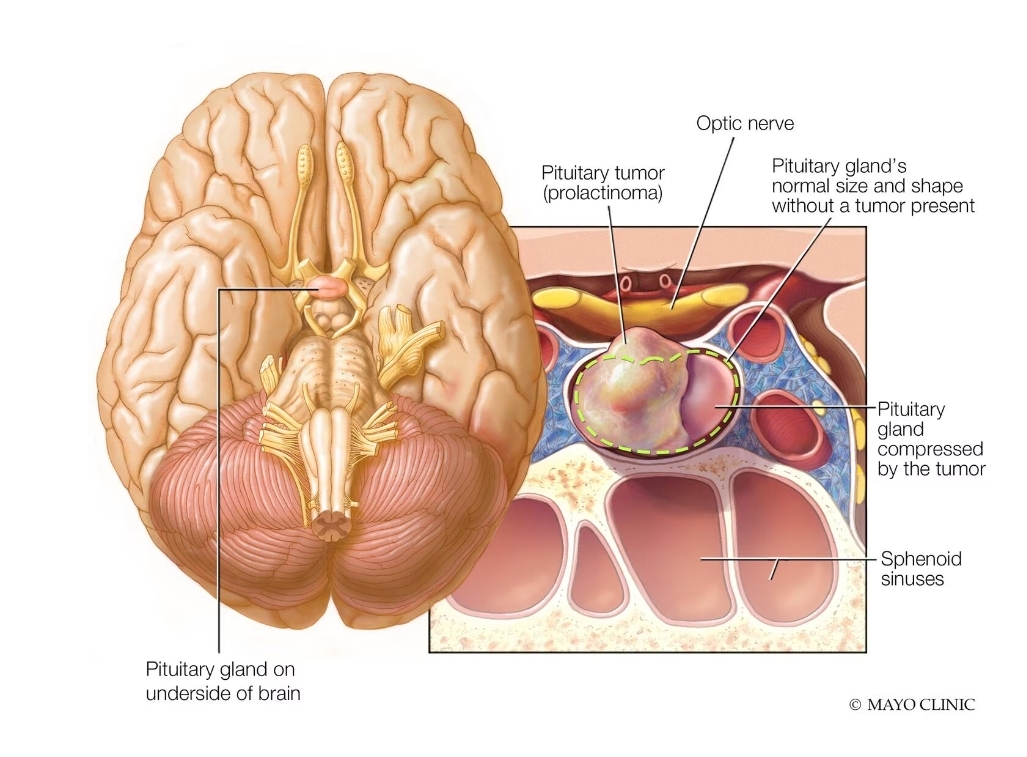
بیماری پرولاکتینوما
معرفی: پرولاکتینوما شایع ترین شکل تومور نورواندوکرین هیپوفیز (PitNET) است که تقریباً نیمی از این تومورها را تشکیل می دهد. آگونیست های دوپامین (DAs) به طور سنتی درمان اولیه برای اکثر پرولاکتینوم ها بوده و جراحی در مرحله بعدی در نظر گرفته می شود.
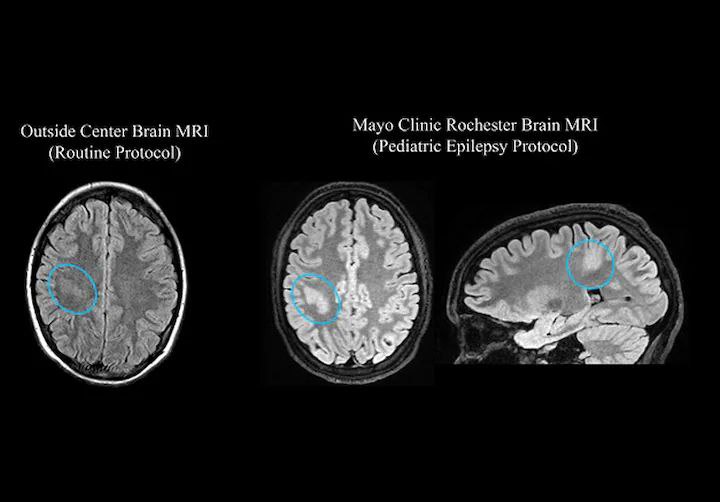
روش کم تهاجمی برای درمان صرع کودکان
مرکز کودکان مایو کلینیک متعهد به استفاده از رویکردهای پیشرفته و کم تهاجم برای صرع کودکان است. مایو کلینیک به عنوان یک مرکز جامع صرع سطح 4 ، طیف وسیعی از گزینه های درمانی را ارائه می دهد که به تمام جنبه های مراقبت از کودکان می پردازد و بیمارانی که به دنبال درمان اولیه یا نظرات دوم هستند از این تجربه و تخصص بهره مند می شوند.